Durch das am 23. Juli 2015 in Kraft getretene GKV Versorgungsstärkungsgesetz (GKV-VSG) werden neue Untersuchungs- und Behandlungsmethoden (NUB) nun einer frühen Nutzenbewertung unterzogen. Das bisherige NUB Anfrageverfahren nach § 6 Abs. 2 KHEntgG wurde somit im Sinne der Patientensicherheit wesentlich erweitert.
Die gesetzlichen Regelungen finden sich im §137 h SGB V. Die Bewertung wird durch den gemeinsamen Bundesausschuss (G-BA) vorgenommen, insofern die Methode den Kriterien unterliegt.
Inhaltsverzeichnis
Ausschlusskriterien
Folgende Kriterien können für einen eindeutigen Ausschluss zum Verfahren der frühen Nutzenbewertung für Medizinprodukte herangezogen werden:
- das Medizinprodukt ist den Klassen I oder IIa zuzuordnen,
- für das Verfahren, für das das Medizinprodukt zum Einsatz kommen soll, wurde vor dem 01.01.2016 eine NUB Anfrage an das InEK versandt,
- das Verfahren / die Methode, weist vor dem 23. Juli 2015 einen spezifischen OPS Code auf,
- es handelt sich um eine reine Weiterentwicklung / Modifikation eines bestehenden etablierten Verfahrens ohne signifikante Veränderung oder Indikationsausweitung = max. Schrittinnovation (kein neues theoretisch wissenschaftliches Konzept)
Prüfungsablauf / Aufnahmekriterien
Erhält der G-BA durch ein Krankenhaus und im Benehmen mit dem Hersteller einen Antrag auf eine frühe Nutzenbewertung, so erfolgt zunächst eine Prüfung der folgenden Merkmale, ob für die angefragte Methode eine Bewertung nach § 137h SGB V durchzuführen ist.
Hierfür prüft der G-BA der Reihe nach, ob für die Methode…
- eine NUB Anfrage erstmalig gestellt wurde (Anfrage nach dem 01.01.2016),
- auf einem Medizinprodukt mit hoher Risikoklasse beruht (näheres hierzu im 2. Kapitel § 30 VerfO 2.4.1)
- ein neues theoretisch-wissenschaftliches Konzept aufweist (näheres hierzu im 2. Kapitel § 31 VerfO)
- den Leistungsanspruch der gesetzlich Krankenversicherten umfasst. (Wirtschaftlichkeitsgebot – § 137c SGB V)
Stellt der G-BA fest, dass es an einer dieser Voraussetzungen fehlt, fällt die gegenständliche Methode nicht in das Bewertungsverfahren gemäß § 137h.
Beschluss / Feststellung aus früher Nutzenbewertung
Sind die Kriterien für eine Bewertung erfüllt, entscheidet der G-BA nach Auswertung der zur Verfügung gestellten unterlagen ob,
1. die Eingliederung in das Leistungsangebot der GKV direkt erfolgen kann,
2. ein Ausschluss aus dem GKV Leistungsangebot ausgesprochen wird (Erlaubnis mit Verbotsvorbehalt erlöscht in diesem Fall)
3. oder ob eine Erprobung nötig ist, um die Evidenz zu vertiefen.
Die Feststellung trifft der G-BA nach Maßgabe der Regelungen des 2. Kapitel §§ 36, 33 Absatz 2 VerfO in Form eines Beschlusses.
Änderungen durch das Terminservice- und Versorgungsgesetz (TSVG)
Das am 11. Mai 2019 in Kraft getretene Terminservice- und Versorgungsgesetz (TSVG) beinhaltet maßgebliche Änderungen zur frühen Nutzenbewertung nach § 137h SGB V sowie der Erprobung von Untersuchungs- und Behandlungsmethoden nach §137e SGB V.
Potentialprüfung entfällt
Die zwischen dem 01. Januar 2016 – 25. März 2020 geltende Potentialprüfung, die bei erkennbarem Potential eine Erprobungsstudie nach sich zog, wurde mit Einführung der Medical Device Regulation (MDR) abgeschafft.
Für den Fall, dass aufgrund der vorgelegten Unterlagen weder
- ein Nutzen noch
- ein Schaden bzw. eine Unwirksamkeit der Methode
als belegt anzusehen ist, erfolgt eine kontrollierte Leistungserbringung im Rahmen der Krankenhausbehandlung.
Es bedarf nicht mehr einer Potentialbewertung und der positiven Feststellung eines Potentials durch den G-BA. Angesichts der Weiterentwicklungen des Konformitätsbewertungsverfahrens nach Maßgabe der Verordnung (EU) 2017/745 (MDR) kann auf das Erfordernis einer positiven Potentialfeststellung als Voraussetzung für eine Erprobung verzichtet werden.
Das Potenzial einer Erprobung ergibt sich ergänzend zu Absatz 3 insbesondere dann, wenn zumindest so aussagefähige wissenschaftliche Unterlagen vorliegen, dass auf dieser Grundlage eine Studie geplant werden kann, die eine Bewertung des Nutzens der Methode auf einem ausreichend sicheren Erkenntnisniveau erlaubt.
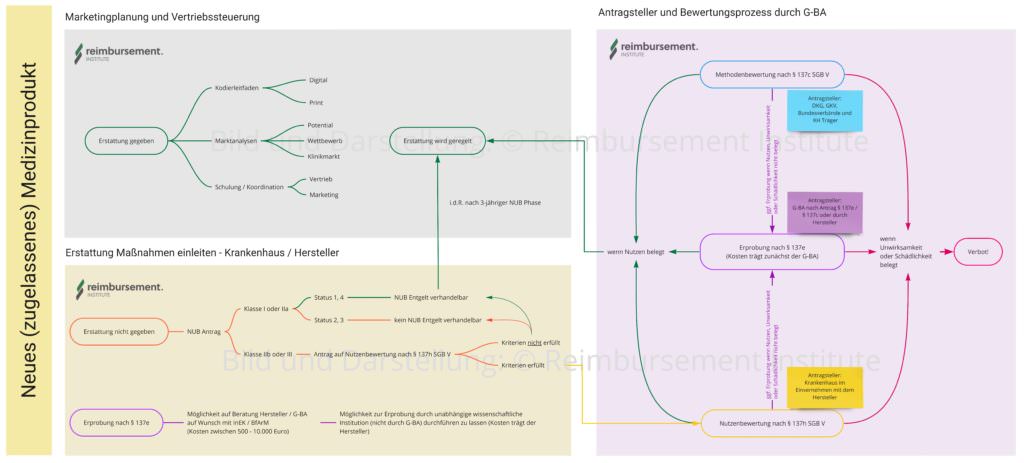
Wege eines zugelassenen Medizinproduktes in die stationäre Erstattung. Analysen, Antragsteller, Verfahrenswege
Beratung wird kostenpflichtig
Der G-BA berät Medizinproduktehersteller zum Verfahren nach § 137h Abs. 6 SGB V. Die Beratung kann auf die Teilnahme des InEK und des BfArM ausgeweitet werden. Gebühren werden ausschließlich auf schriftliche Anforderung einer Beratungsleistung erhoben.
Für die Entscheidung, ob eine Methode dem Verfahren nach § 33 (Prüfung der Voraussetzungen einer Bewertung nach § 137h Absatz 1 Satz 4 SGB V) Absatz 1 VerfO unterfällt, fallen keine Gebühren an. Für die Fälle, bei denen Beratungsleistungen hinzukommen, fallen die folgenden Gebühren an.
825,00 € für allgemeine Anfragen zu:
- den formalen Voraussetzungen der Informationsübermittlungspflicht nach § 137h Absatz 1 Satz 1 SGB V,
- den Voraussetzungen der Erbringung einer Untersuchungs- oder Behandlungsmethode zu Lasten der Krankenkassen.
3.300,00 € für allgemeine Anfragen zu:
- den Voraussetzungen und dem Verfahren zur Bewertung von NUBs mit Medizinprodukten hoher Risikoklasse nach § 137h Absatz 1 Satz 4 SGB V,
- dem Verfahren einer Erprobung sowie zu den Alternativen der Finanzierung der wissenschaftlichen Begleitung und Auswertung der Erprobung nach § 137e Absatz 5 Satz 2 SGB V,
- oder zu im Aufwand vergleichbaren sonstigen Anfragen.
11.500,00 € für Anfragen mit unmittelbarem Bezug zu einer konkreten Untersuchungs- und Behandlungsmethode (bzw. Medizinprodukt) zu:
- verfahrenstechnischen und methodischen Anforderungen an die Bewertung einer Untersuchungs- und Behandlungsmethode und den Voraussetzungen des Verfahrens zur Bewertung von NUBs mit Medizinprodukten hoher
Risikoklasse nach § 137h Absatz 1 Satz 4 SGB V, - den Voraussetzungen und dem Verfahren zur Finanzierung der Erprobung durch den G-BA nach § 137e Absatz 6 SGB V oder Selbstdurchführung nach § 137e Absatz 5 Satz 2 SGB V,
- oder zu im Aufwand vergleichbaren sonstigen Anfragen.
16.500,00 € für Anfragen zu:
- den verfahrenstechnischen und methodischen Anforderungen an die Bewertung einer konkreten Untersuchungs- und Behandlungsmethode unter Berücksichtigung der betroffenen Zielpopulation, der zweckmäßigen (angemessenen) Vergleichstherapie sowie der patientenrelevanten Endpunkte einschließlich der Voraussetzungen einer entsprechenden Erprobung,
- oder zu im Aufwand vergleichbaren sonstigen Anfragen
hier geht´s zur Gebührenordnung
Kostenübernahme für Erprobung geregelt
Medizinproduktehersteller oder Unternehmen, die ein wirtschaftliches Interesse an einer Erbringung zulasten der Krankenkassen haben, haben künftig das Wahlrecht, die wissenschaftliche Begleitung und Auswertung der Erprobung statt durch den GBA unmittelbar selbst und auf eigene Kosten zu beauftragen. Die Vorgaben des G-BA sind einzuhalten.
-> Beauftragung zur Erprobung durch G-BA – Kosten werden vom G-BA getragen aus dem Systemzuschlag
-> Beauftragung zur Erprobung durch Unternehmen – Kosten sind von diesem zu tragen
Beruht die zu erprobende NUB auf dem Einsatz eines Medizinproduktes und die Beauftragung erfolgt durch den G-BA, wird eine Vorfinanzierung der gesamten Kosten durch den G-BA vorgenommen, um den zeitnahen Beginn der Erprobung und den für die Aufgabenwahrnehmung des G-BA notwendigen Erkenntnisgewinn zu befördern.
Erst nach Abschluss der Erprobung erfolgt eine nachträgliche Kostenbeteiligung der betroffenen Medizinproduktehersteller. Nur wenn es aufgrund der Erprobung tatsächlich zu einer Aufnahme der NUB in die ambulante Regelversorgung nach § 135 kommt.
Weitere, relevante Informationen:

